El caso de Sare y Cornelia de Lange
Se llama Sare y tiene cuatro años. Esta niña es uno de los cuatro casos diagnosticados en Euskadi del síndrome de Cornelia de Lange. Hay 70 casos conocidos en España y la incidencia general es de uno de 10.000-100.000 nacimientos, cifra que está en debate. Es de 0,97 por cada 100.000 nacimientos en España. En una revisión de los enfermos con síndrome de Cornelia de Lange, conocido como CdLS por sus siglas en inglés, con datos de Europa entre 1980 y 2002 y publicada en 2008 por Ingeborg Barisic y sus colegas, de la Universidad de Zagreb, según 33 bases de datos de 16 países con más de ocho millones de nacimientos, encontraron 106 enfermos y, por tanto, una incidencia de 1 enfermo por 81.000 nacimientos. La incidencia es baja pero la cifra real no se conoce con exactitud.
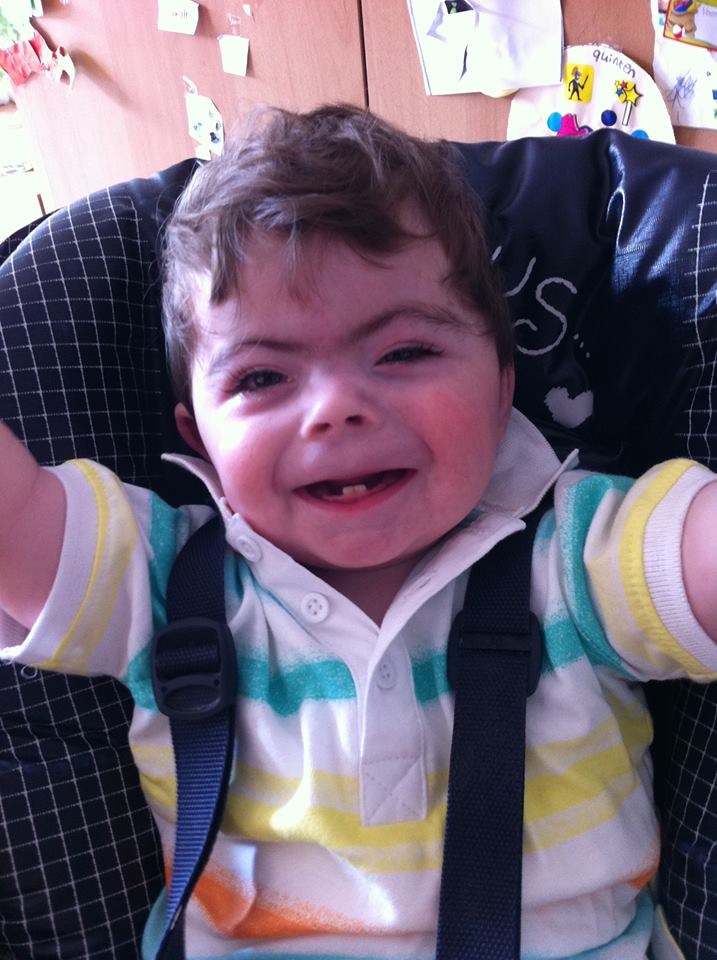
Imagen: Wikimedia Commons.
Sare nació con 1,5 kilos de peso y, ahora, con cuatro años, pesa 7 kilos. Como su desarrollo es lento, pronto empezará a andar y no habla, aunque se comunica con gestos y por contacto visual. Según cuenta su madre, Katia, es una niña que sabe lo que quiere y lucha por conseguirlo, persevera en ello y trabaja duro. Va a la escuela dos días a la semana y le encanta la música en sus sesiones de musicoterapia.
El síndrome de Cornelia de Lange es una alteración genética que afecta al desarrollo físico e intelectual del paciente y presenta unos rasgos físicos típicos, aunque hay pacientes que no presentan los síntomas característicos. Los cambios se deben mutaciones espontáneas en los cromosomas en el 99% de los casos, o por herencia de ambos progenitores en un porcentaje muy bajo. Es, por tanto, un trastorno malformativo múltiple congénito. Se determina por sus características faciales en asociación con retraso en el crecimiento pre- y postnatal, retraso intelectual de nivel variable y, en algunos casos, anomalías en las extremidades superiores. Muchos de estos síntomas aparecen en el nacimiento y a una edad temprana.
Para facilitar el diagnóstico del síndrome, es importante detallar una descripción de los síntomas y, así, conseguir la detección temprana y el inicio del tratamiento de los niños. Las características del CdLS más habituales son el bajo peso al nacer, crecimiento lento, baja estatura y microcefalia. En el rostro destacan cejas unidas en el centro y pestañas largas, nariz pequeña y respingona, y labios finos en V invertida. Suele haber hirsutismo con pelo excesivo, manos y pies pequeños, unión parcial del segundo y tercer dedo de los pies, dedos meñiques curvados, reflujo gastroesofágico, problemas cardíacos, fisura del paladar, anormalidades intestinales, dificultad en la alimentación y pérdida de audición y visión. Todos estos síntomas aparecen con una enorme variabilidad según cada paciente.
La expectativa de vida de los pacientes con el síndrome Cornelia de Lange no se conoce con exactitud. Hasta hace unos años, por la falta de un diagnóstico temprano y acertado y de conocimiento sobre el síndrome, la mortalidad a corta edad era elevada pero, en la actualidad, con los cuidados médicos adecuados y un buen ambiente familiar, pueden llegar a adultos. Leah Dowsett y su grupo, del Hospital Infantil de Filadelfia, después de revisar 246 enfermos de quince países, menciona que alguno de los pacientes había llegado a los 37 años. En cuanto al nivel intelectual, hay casos excepcionales como el que menciona Juan Pié, de la Universidad de Zaragoza, de una paciente que sacó el carnet de conducir.
Hay tres tipos aceptados de CdLS que son, según M. C. Gil, de la Universidad de Zaragoza: tipo I, el más leve, con signos faciales menos marcados o más tardíos, crecimiento casi normal y retraso mental leve o inexistente; tipo II, moderado, con mucha variabilidad, rasgos faciales característicos pero parciales, habla y comunicación limitadas, crecimiento más o menos la mitad de lo normal; y tipo III, el más grave, o forma clásica, con crecimiento lento en el útero, graves retrasos y malformaciones, pérdida significativa de la comunicación y discapacidad severa.
En 1933, la pediatra holandesa Cornelia de Lange describía el síndrome en dos niñas con rasgos físicos similares y, en la actualidad, se acepta que son los síntomas de la enfermedad que ahora conocemos con su nombre. El primer caso fue una niña con 14 meses de edad que pesaba 5,5 kilos y había nacido con 1,250 kilos. Estuvo ingresada en el hospital 42 días. El segundo caso, también una niña, tenía 5 meses de edad y un peso de 3,5 kilos y había nacido con 2,0 kilos.

Cornelia de Lange había nacido el 24 de junio de 1871 en Alkmaar, pequeña ciudad al norte de Amsterdam. Murió el 28 de enero de 1950 en Amsterdam. Su padre era abogado y miembro del Parlamento holandés. Su hija Cornelia fue la primera mujer de Alkmaar que terminó el bachillerato superior.
Quiso ir a la universidad pero su padre opinaba que, antes, debía ir a Suiza un año “para terminar su educación de una manera más acorde a una mujer de su estatus social y época”. Cuando volvió a Alkmaar seguía queriendo ir a la universidad y, en 1891, consiguió el permiso de su padre y se matriculó en química, disciplina que, entonces, se consideraba adecuada para una joven. Al año siguiente se pasó a medicina y se graduó en 1897. Ese mismo año escribió la tesis con el título “Análisis comparativo de restos humanos”. Comparaba la composición química de la leche con la de fetos humanos. Fue la cuarta mujer en Holanda en defender una tesis en medicina.
Pero la pediatría, que era la especialidad que le interesaba, no se impartía en Holanda y, por ello, se trasladó a Suiza, al Hospital Infantil de Zurich. Pocos años después volvió a Holanda y se integró, en 1907, en el Hospital Infantil de Amsterdam. Estableció una consulta para mujeres y niños y pronto fue conocida por su buen trabajo. Estudió anatomía y neurología y se interesó por la anatomía patológica y por la práctica de autopsias. Fue pionera en muchos campos de la medicina, especialmente en pediatría clínica, neuropediatría y genética. En esos años, cuando Cornelia de Lange ya ejercía y empezaba a ser conocida como pediatra, un tío suyo, también médico le escribió una carta que, entre otras cosas, decía, según Miguel Ángel Zafra y sus colegas, del Hospital Universitario de Fuenlabrada, “Querida Cornelia: ha sido terrible para la familia que estudiaras para ser doctor pero, ahora, además, publicas sobre infección urinaria en lactantes. Verdaderamente, has llegado demasiado lejos”.
Sin embargo, era conocida por su trato amable y tranquilo con los pacientes. Era modesta y sobria pero, también, cálida y con gran simpatía. En 1932 escribió que “yo creo que el pediatra debe ser capaz de vivir alegre como un niño. Debe revolotear con las mariposas, juguetear con borreguitos, arrullarse con los bebés, y conservar en lo más profundo de su ser algo de la eterna juventud”.
En 1927, a los 57 años, ganó una plaza de pediatría en la Universidad de Amsterdam y, así, fue la primera mujer profesora de medicina en Holanda. En su puesto dirigió catorce tesis en pediatría y, entre sus graduados, había tres mujeres. Fue en estos años, en 1933, cuando describió los dos primeros casos del síndrome que lleva su nombre. Años más tarde, en 1938, publicó un estudió con un tercer caso del síndrome y los hallazgos de la autopsia de su primer caso pues la paciente había fallecido con 5 años y 9 meses de edad.
Cuando se retiró, en 1945, al acabar la Segunda Guerra Mundial, le cedieron en el hospital un pequeño despacho y un laboratorio y allí, hasta su muerte en 1950, desarrolló una gran actividad como consultora para sus colegas.
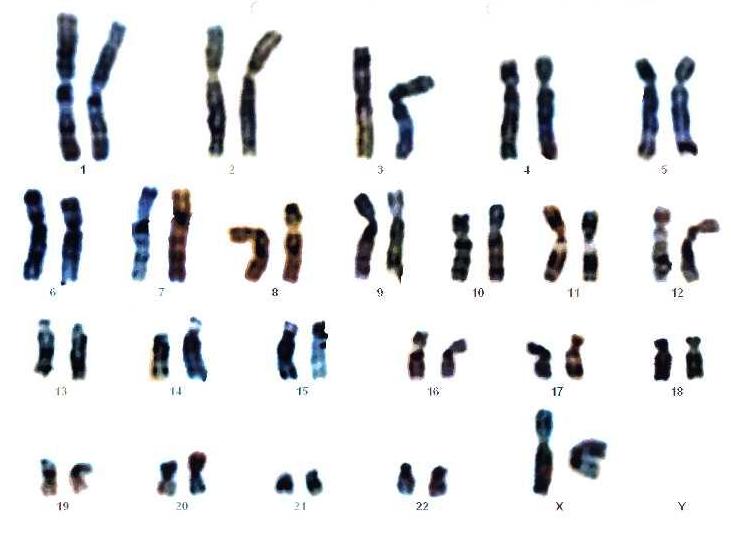
Imagen: Wikimedia Commons.
Desde 1938 se habían descrito otros casos en Europa y, en 1963, se publicó el primero en Estados Unidos. El primer paciente en España fue descrito en 1961 por P. Altozano, de la Residencia Sanitaria del SOE en Cáceres. Era una niña, llamada Encarnación, natural de Cáceres y de 5 meses de edad. La descripción y el estudio de P. Altozano es muy detallada y el diagnóstico acertado. Encarnación nació con 1,650 kilos de peso y, a los 5 meses, pesaba 3,430 kilos.
En 2004, el grupo de Ian Krantz, del Hospital Infantil de Filadelfia, encontró el primer gen mutado causante de más de la mitad de los casos conocidos del síndrome. Investigaron doce familias con casos de CdLS y detectaron un gen del cromosoma 5 al que nombran NIPBL y que aparece mutado en seis de las familias.
Poco después, en 2006, un grupo liderado por Antonio Musio, del Instituto de Tecnologías Biomédicas de Segrate, en Italia, encontró otro gen mutado relacionado con el síndrome. Trabajaron con 57 pacientes con diagnóstico de CdLS. En 24 de ellos encuentran la mutación del gen NIPBL, descrito por el equipo de Philadelphia. En una familia de los 33 pacientes restantes encuentran, en el cromosoma X, una mutación del gen SMC1A relacionada con el síndrome. Es la segunda mutación que interviene en el CdLS.
Al año siguiente, en 2007, el grupo de Filadelfia, liderado por Matthew Deardorff, encontró un tercer gen mutado relacionado con el CdLS. Aparece en el cromosoma 10 y se denomina SMC3. Es notable que se ha encontrado una mutación en este gen en un paciente autista.
Y, ya en 2012, Matthew Deardorff y su equipo, del Hospital Infantil de Filadelfia, localizaron otros dos genes mutados relacionados con el síndrome. Eran el RAD21, del cromosoma 8, y el HDAC8, del cromosoma X. Como los tres primeros, también intervenían en el complejo de las cohesinas.
La mutación más extendida es la que afecta al gen NIPBL, en el 80% de los pacientes, seguida de la que afecta al SMC1A en el 15%. Esta última apareció después de estudiar a 100 pacientes y no se han encontrado nuevos casos. Por tanto, tiene una incidencia muy baja. Los casos más graves aparecen con la mutación en el NIPBL, seguido por HDAC8, RAD21, SMC1A y SMC3 en los enfermos con el síndrome más leve.
Hay que destacar que casi el 35% de los pacientes diagnosticados con el CdLS no tienen mutaciones en ninguno de los genes encontrados hasta ahora. Este hecho implica que puede haber otros genes mutados todavía no identificados.
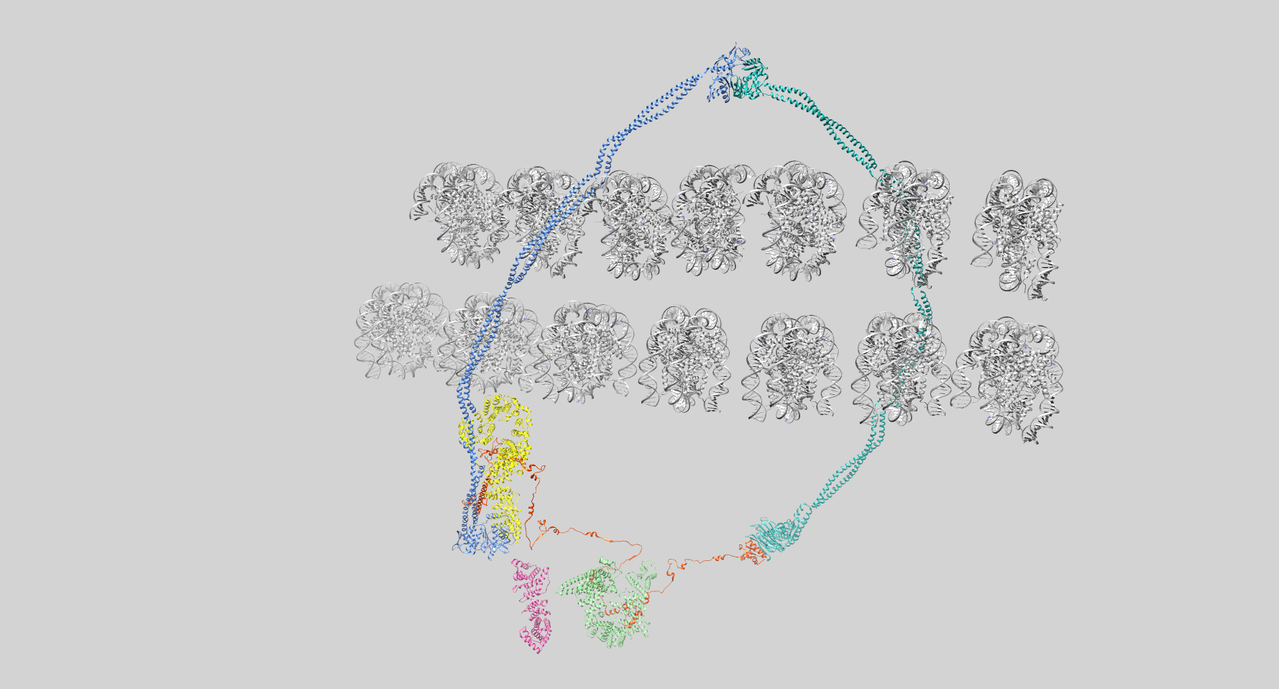
Todos estos genes codifican proteínas relacionadas con las cohesinas. Como revisa Erez Lieberman Aiden, de la Universidad de Harvard, son un elemento fundamental de la estructura y cohesión de los cromosomas durante la duplicación del ADN, en su reparación, y en su funcionamiento, y, también, en la separación de las cromátidas, o nuevos cromosomas, después de la duplicación. Por tanto, las mutaciones en los genes encargados de las cohesinas interfieren en el buen funcionamiento del ADN y puede que no regulen bien la expresión de genes y no mantengan la estructura correcta de los cromosomas.
Cuando la preguntaron a Ana Losada, ahora en el Centro Nacional de Investigación Oncológica en Madrid, pero en los noventa en el Laboratorio de Cold Spring Harbor de Nueva York donde, con el grupo de Tatsuya Hirano descubrió las cohesinas, cuántos genes podían estar implicados en el síndrome de Cornelia de Lange, respondió que una cincuentena pues el control génico de las cohesinas es muy complejo. Da una idea de lo que queda por trabajar en el campo de las enfermedades ahora llamadas cohesinopatías.
Por otra parte, hay varios tipos de cáncer que muestran mutaciones en los mismos genes que el CdLS. La hipótesis de investigación planteada es que, si las mutaciones afectan a la función de mantener la cohesión entre cromosomas, aparece cáncer, y, si afecta a la expresión de los genes, aparece el síndrome.
Y no olvidemos a Sare. Cuanto más se conozca el síndrome, mejores diagnósticos se conseguirán y más investigación se dará a conocer. Así entenderemos mejor a niños como Sare que, y su madre Katia nos lo cuenta, “con Sare es el día a día, no sabemos nada … que llegue hasta donde llegue, si es que tiene un límite… que logre la mayor autonomía posible, pero sin agobiarnos si es una niña que no consigue autonomía”.
Referencias
- Aiden, E.L. 2019. Desenredar el genoma. Investigación y Ciencia mayo: 32-38
- Altozano, P. 1961. Un caso de nanismo typus amstelodamensis. Revista Española de Pediatría 17: 319-332
- Síndrome de Cornelia de Lange. Wikipedia 20 diciembre 2018
- Cornelia Catharina de Lange. Wikipedia 11 marzo 2019
- Barisic, I. et al. 2008. Descriptive epidemiology of Cornelia de Lange Syndrome in Europe. American Journal of Medical Genetics 146: 51-59
- Deardorff, M.A. et al. 2007. Mutations in cohesin complex members SMC3 y SMC1A cause a mild variant of Cornelia de Lange síndrome with predominant mental retardation. American Journal of Human Genetics 80: 485-494
- Deardorff, M.A. et al. 2007. RAD21 mutations cause a human cohesinopathy. American Journal of Human Genetics 90: 1014-1027
- Deardorff, M.A. et al. 2012. HDAC8 mutations in Cornelia de Lange Syndrome affect cohesin acetylation cycle. Nature 489: 313-317
- Dowsett, L. et al. 2019. Cornelia de Lange Syndrome in diverse populations. American Journal of Medical Genetics DOI: 10.1002/ajmg.a.61033
- Gil, M.C. et al. 2010. Síndrome de Cornelia de Lange. Asociación Española de Pediatría Protocolos 1: 1-10
- Krantz, I.D. et al. 2004. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drsophila melanogaster Nipped-B. Nature Genetics 36: 631-635
- de Lange, C. 1933. Sur un type nouveau de dégénération (typus Amstelodamensis). Archives Médecine Enfants 36: 713-719
- de Lange, C. 1938. Nouvelle observation du “Typus Amstelodamensis” et examen anatomopathologique de ce type. Archives Médecine Enfants 41: 193-203
- Losada, A. et al. 1998. Identification of Xenopus SMC protein complexes required for sister crhromatid cohesion. Genes & Development 12: 1986-1997
- Martínez, M. 2018. Una luchadora llamada Sare. Deia 23 diciembre
- Musio, A. et al. 2006. X-linked Cornelia de Lange síndrome owing to SMC1L1 mutations. Nature Genetics 38: 528-530
- Pié Juste, J. 2014. Síndrome Cornelia de Lange: Investigación en tránsito. Discurso de Recepción de la Real Academia de Medicina de Zaragoza. 42 pp
- Zafra, M.A. et al. 2014. Síndrome de Cornelia de Lange. Canarias Pediátrica 38: 36-41
Sobre el autor
Eduardo Angulo es doctor en biología, profesor de biología celular de la UPV/EHU retirado y divulgador científico. Ha publicado varios libros y es autor de La biología estupenda.
6 comentarios
Soy madre de una niña de 18 años con Cornelia de Lange en estado grave. Muy buen informe, completo y claro. Ahora se conoce mucho más sobre Síndrome. Gracias por compartir la información.
Gracias por el comentario y, realmente, una de las razones fue dar a conocer el síndrome y contribuir a su investigación. Un abrazo muy fuerte para tu niña y otro lleno de ánimo y de fuerza para ti.
Soy tia de una niña de 23 años con este sindrome.Gracias por compartir la información.
Buenas . Hola , soy madre de un bebé de 2 años y 6 meses que tiene el síndrome Cornelia y me a ayudado bastante la información dada en esta página .muchas gracias ..
Me alegro de que te haya sido útil, es uno de los objetivos. Además, deben conocerse y asociarse las familias para conseguir más apoyos y, sobre todo, más investigación.
Hola , tengo 3 hijas la última de 12 años afectada por el síndrome , al principio no me daban esperanzas de vida por tantas dificultades que tenía , me alienta saber que se va conociendo más el síndrome y que las investigaciones nos ayuden a continuar con todas las variables y múltiples condiciones a las que nos enfrentamos con estos pequeños